


近期,磁共振中心王为教授研究团队结合多种表征手段(ssNMR、GC-MS 分析)与密度泛函理论(density functional theory,DFT)计算,揭示了H-Y分子筛上乙烯转化初期的反应机制。该研究成果发表于《催化科学与技术》(Catalysis Science & Technology Catalysis Science & Technology)上。
H-Y 分子筛上乙烯转化初期的反应机制
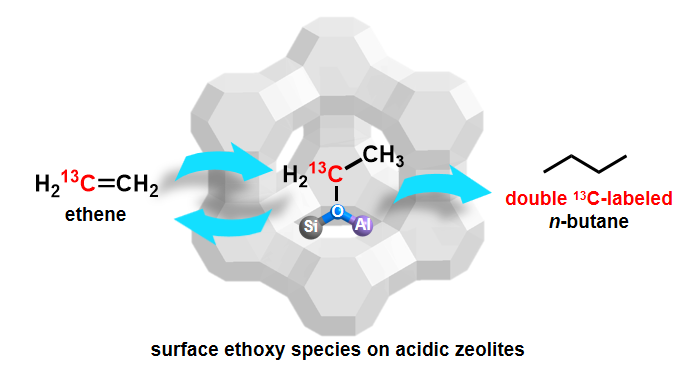
乙烯是石油化工产业中最重要的基础原料之一,分子筛催化的乙烯二聚/低聚过程能够将乙烯转化为高附加值的产品,该过程在学术界和工业界引起了的广泛关注。然而,乙烯在酸性分子筛上的快速低聚严重阻碍了对反应中间体的观测和分析,目前仍缺乏对其反应机理的深入理解。在前期工作基础上(Angew. Chem. Int. Ed., 2015, 54, 7363.; Acc. Chem. Res., 2008, 41, 895; J. Am. Chem. Soc. 2006, 128, 11679),借助选择性同位素标记和ssNMR,首次在分子筛上观测到乙烯转化为表面乙氧基物种。通过分析13C同位素示踪实验中产物的同位素分布情况,鉴定了表面乙氧基物种反应的初始C4产物为正丁烷。在此基础上,结合DFT理论计算,进一步确认了表面乙氧基物种在C‒C键形成过程的中间体作用。这一结果不仅阐明了表面乙氧基物种的反应活性,而且揭示了乙烯转化过程中初始C‒C键的形成机理。
该工作得到了国家自然科学基金项目(No. 21903041)、甘肃省教育厅科研计划项目(No. 2021jyjbgs-04)和中央高校基本科研业务费专项基金项目(No. lzujbky-2021-ey11)资助。
 当前位置:
当前位置: